US FDA Class III Medical Device – PMA
Simplified Roadmap
QMS-SR-US-FDA-MD(PMA)-RC3
Version: 07/18/2025
Prepared by: Dr. Vlad Reznikov
Date: July 18, 2025
Executive Summary
Class III devices require Premarket Approval (PMA). A Q‑Sub (Pre‑Submission) phase is essential to align on clinical and non‑clinical study expectations.
Estimated Timeline
≈ 3–7 years total, driven largely by clinical‑study duration.
Key Documents Checklist
| Document | Reference |
| Q‑Sub Packages (Pre‑Subs) | FDA Q‑Sub Guidance |
| IDE Application | 21 CFR 812 |
| Clinical Study Reports | ISO 14155 / GCP |
| PMA Volumes 1–4 | 21 CFR 814 |
| Process Validation & Sterility | 21 CFR 820 |
| Post‑Approval Study Protocol | PMA Order Conditions |
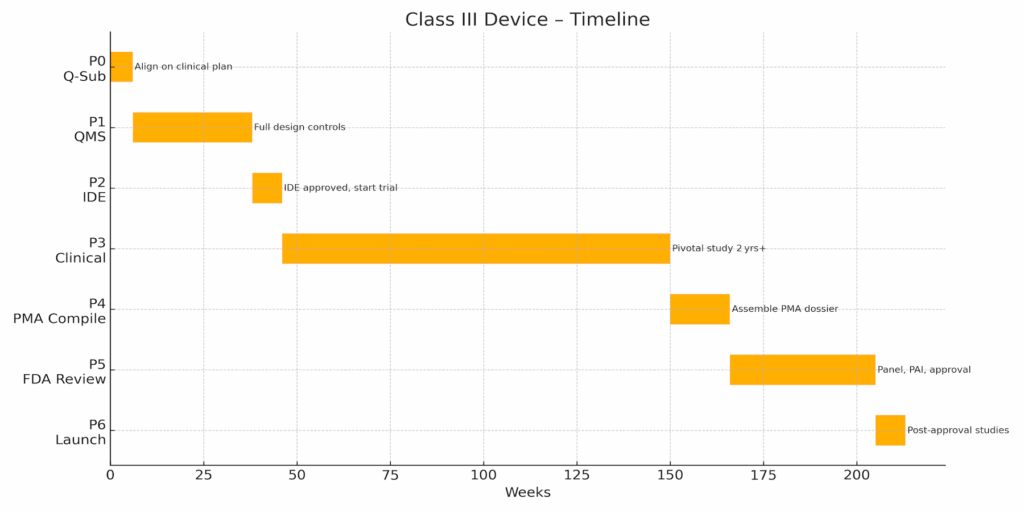
Phase 0 – Strategy & Initial Q‑Sub (≈6 wks)
- Define intended use; hold Pre‑Sub to discuss clinical pathway & endpoints.
- Outline IDE requirements; set regulatory project plan.
Phase 1 – QMS Expansion & Design Controls (≈32 wks)
- Establish full ISO 13485 / 21 CFR 820 QMS with design controls.
- Compile DHF, DMR, process validation, human‑factors protocols.
Phase 2 – IDE Submission & Approval (≈8 wks)
- Prepare IDE dossier; obtain IRB & FDA approvals.
- Site initiation and training.
Phase 3 – Pivotal Clinical Investigation (≈104 wks)
- Conduct multi‑site trial; periodic IDE reports.
- Interim analyses and risk monitoring.
Phase 4 – PMA Compilation (≈16 wks)
- Assemble PMA Volumes 1–4, draft summaries, pay user fee.
- Pre‑Submission meeting for PMA filing strategy (optional).
Phase 5 – FDA Review & Panel (≈39 wks)
- Address major/minor deficiencies; support advisory panel if convened.
- Undergo Pre‑Approval Inspection (PAI).
Phase 6 – Approval & Launch (≈8 wks, then ongoing)
- Fulfill post‑approval study commitments; submit PMA annual report.
- Maintain heightened MDR and vigilance.