United States of America – Radiopharmaceuticals & Contrast Agents – Radiopharma Authorization
Simplified Roadmap
QMS-SR-RA-FDA-radiopharma-authorizatio
Version: 08/20/2025
Prepared by: Dr. Vlad Reznikov
Date: August 19, 2025
Executive Summary
This roadmap describes Radiopharma Authorization in United States of America for the Radiopharmaceuticals & Contrast Agents category. Consider market size, population, GDP and PPP when prioritizing launch order—see GDP Matrix (ROCKSTARS, HONEYBEES, MAVERICKS, UNDERDOGS): https://patternofusa.com/gdpmatrix/. Peer markets with similar frameworks may allow reliance or abridgement where eligible.
Key Regulations & Requirements
National Regulatory Authority (NRA): FDA
Documents / Standards / Certifications:
- CMC with isotope controls
- Sterility/endotoxin validation
- Short shelf-life SOPs
- Clinical evidence
- Radiation precautions labeling
- Site licensure
Notes: Submission format, portal, legalization, and language for United States of America require verification .
Estimated Timeline
Zero‑based Gantt timeline (weeks). Bars are illustrative; adjust per project scope.
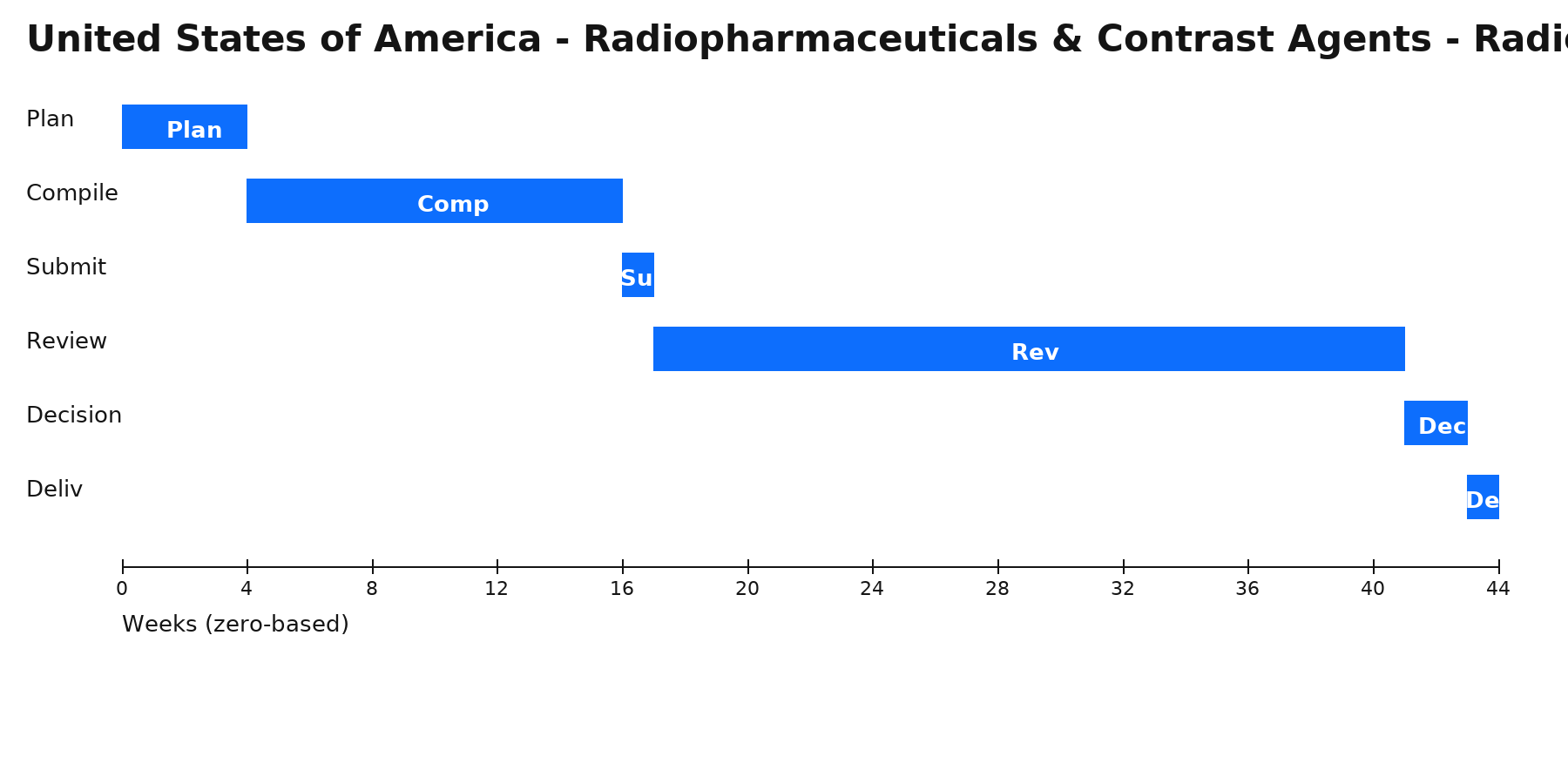
Phases & Tasks
Plan (4 wks) — Define pathway, reliance, local roles (MAH/agent); build high-level plan & risk register.
Compile (12 wks) — Assemble dossier/quality evidence; confirm translations, legalization/apostille, and samples/testing.
Submit (1 wk) — File via portal/gateway; complete fee payment and resolve validation issues.
Review (24 wks) — Manage queries/clock-stops; submit clarifications, addenda, or product samples as requested.
Decision (2 wks) — Receive authorization or additional requests; align launch, PV, and supply readiness.
Deliv (1 wk) — Issue internal release pack (certificates, approved labels, SOPs) and plan for renewals/variations.
Deliverables
- Authorization/Certificate or Registration Record
- Approved labeling/artwork (as applicable)
- Final dossier, correspondence log, and post‑approval plan